A continuación se va a dar una visión del Síndrome X Frágil
en los siguientes aspectos:
Introducción
Causas
del Síndrome
Como
se produce
Como
influye el sexo de las personas
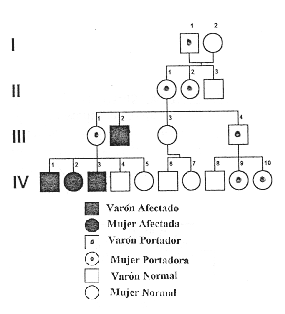
Ejemplo
genealógico
Como
se detecta
Incidencia
Problemática
y efectos
Rasgos
físicos
Comportamiento
Tratamiento
Tratamiento
psicopedagógico y conductual
Conclusiones
finales
Bibliografía
Glosario
Anexo
1 - Prolapso de la válvula mitral
Anexo
2 - Estado de las investigaciones
Anexo
3 - Intervención multidisciplinar
| Puede descargar una copia
de este informe pulsando aquí |
 |
El presente informe ha sido realizado por la
Asociación Síndrome X Frágil de Madrid, inscrita en el
Registro de Asociaciones de la Comunidad de Madrid con el número
17.011.
Este trabajo es producto de la compilación de la documentación
disponible por la Asociación. Los términos, descripciones,
conceptos, consejos, proceden de dicha documentación.
Un síndrome, en medicina, es un conjunto
de signos y síntomas que existen al mismo tiempo y que definen
clínicamente un estado de enfermedad. En el caso del síndrome X-Frágil,
la causa de estos síntomas viene dada por una anomalía en un
cromosoma sexual X.
Un tipo especial de retraso mental hereditario ligado al sexo ya
fue descrito en los años cuarenta por Martin y Bell (1943) y
posteriormente por Renpenning et al.(1962), a partir del estudio
clínico de familias con diversos casos de retraso mental en
varones.
El correlativo citogenético de esta enfermedad, descubierto por
Lubs en 1969, fue definido como una fragilidad en el brazo largo
del cromosoma X. El actual nombre de síndrome X-Frágil lo
introdujo diez años después G.R. Sutherland (1979).

Cada persona posee 23 pares de cromosomas. Una
de estas parejas determina el sexo con el que se nace, adoptando
el nombre de "cromosomas sexuales". Por su forma se
identifican los cromosomas sexuales femeninos (determinan que la
persona sea de sexo femenino) como XX, y la pareja de cromosomas
masculinos como XY (determinan que la persona sea de sexo
masculino).
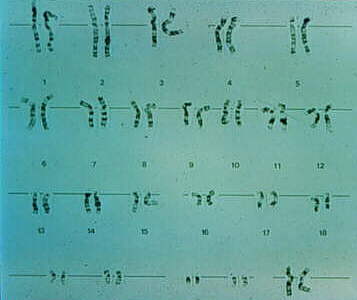
A continuación se muestra la figura de un cariotipo de una
mujer, identificado por los dos cromosomas X en la esquina
inferior derecha.
Por tanto, las mujeres pueden tener esta anomalía
en cualquiera de los dos cromosomas sexuales X, mientras que los
hombres pueden padecerlo sólo en el único cromosoma sexual X
que poseen.
La anomalía es debida a una mutación genética del ADN que
afecta tanto a las células sexuales (óvulos y espermatozoides)
como a los otros tipos de células de nuestro organismo.
Se sabe en la actualidad que esta mutación es producida
inicialmente por el exceso de repetición de una tripleta de
bases nitrogenadas: concretamente la CGG (Citosina, Guanina,
Guanina). Ello hace que se produzca en exceso lo que en química
se llama grupos de metilo (se produce una hipermetilación en la
zona llamada "isla CpG"), dañando principalmente al
gen situado en el locus Xq.27.3 (final del brazo largo del
cromosoma X), que está junto al locus afectado de la
hipermetilazión, influyendo definitivamente también a la proteína
(cromatina) que envuelve al cromosoma X que en este locus se ve
disminuida haciendo más frágil al cromosoma. Este gen se ve
anulado y no puede ejercer su función, fabricar la proteína
llamada FMR-1-P, que ha sido identificada en diferentes tejidos,
sobre todo las neuronas, y de la cual se sabe que juega un papel
importante en el normal desarrollo del cerebro.
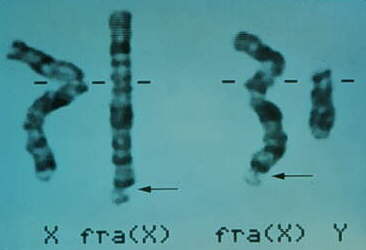
En la imagen siguiente se indica el lugar donde se localiza el
gen FMR-1 (Fragile X Mental Retardation) (gen del retraso mental
por X frágil).
La mutación del cromosoma sexual X no se
produce de golpe, sino que suele seguir un proceso que puede
abarcar varias generaciones de una misma familia.
La mejor manera de explicar el proceso es partir del número de
repeticiones de la tripleta CGG. Atendiendo a este criterio,
podemos encontrar tres posibles estados del cromosoma
- Normalidad: un cromosoma X no afectado
suele presentar entre 5 y 50 repeticiones de la tripleta
CGG en el locus en cuestión.
- Premutación o predisposición: las
repeticiones suelen estar entre 50 y 200, permitiendo al
gen ser aún funcional y fabricar la proteína que le
corresponde. En este caso se habla de mujeres portadoras
y hombres transmisores normales.
- Mutación completa: en este caso las
repeticiones son más de 200 y pueden llegar a varios
miles.
La mutación o las repeticiones de la tripleta
CGG se modifican cuando se transmiten de padres a hijos y tienden
a aumentar en mayor medida cuando la premutación pasa a través
de la mujer. Este cambio explica la mutación del cromosoma X frágil
que se puede encontrar en una persona intelectualmente normal.
Actualmente se sabe que el estado de premutación es inestable
durante la formación de la célula germinal femenina (óvulo),
expandiéndose a mutación completa en la siguiente generación
donde pueden nacer hijos afectados.
En el caso de los varones, la premutación es estable en la
formación de espermatozoides (espermatogénesis) y permanece
como tal en sus hijas, que siempre son normales; pero éstas podrían
tener hijos afectados en los que la premutación se expandiría a
mutación completa (es el fenómeno llamado "anticipación
genética" o "paradoja de Sherman").
Existen diferencias importantes derivadas del
sexo en dos aspectos principales:
- Afectación: como los cromosomas sexuales
femeninos son XX, las mujeres tienen una defensa
adicional importante que provoca que se vean menos
afectadas: si uno de los cromosomas X tiene la mutación,
siempre tienen el otro cromosoma X que puede suplir y
tapar la anomalía de su par. En cambio, los hombres
tienen un solo cromosoma X (el otro es el Y), por lo que
la mutación en el cromosoma sexual X no puede ser
suplida por ningún otro, y la afectación será casi
segura.
- Herencia: las consecuencias de estar
afectados respecto a los descendientes son diferentes según
sea el padre o la madre quien sea el portador. El padre
portador puede transmitir el cromosoma X afectado a sus
hijas pero nunca a sus hijos, pues a éstos les
transfiere el cromosoma Y. La madre portadora, tiene la
probabilidad del 50% de transmitir el gen frágil X a
cada uno de sus hijos o hijas.
El test de laboratorio más frecuente usado ha
sido el análisis citogenético, llamado también análisis
cromosómico, que normalmente se efectúa con células de
muestras de sangre (linfocitos). En ella se puede obtener una
imagen del cromosoma X más o menos nítida y observar si existe
un punto frágil o una rotura en el locus estudiado (Xq27.3).
Esta característica no se puede observar en todas las células,
sino que sólo se ve entre un 4 y un 50% de las estudiadas. En
todo caso, mediante este análisis no se pueden observar los
genes y, por lo tanto, es muy difícil apreciar los estados de
premutación, por lo que no se detectará en la mayoría de las
mujeres portadoras y los varones transmisores normales.
Posteriormente surgió la técnica de los estudios de enlaces de
ADN, pero requerían análisis sanguíneos de múltiples miembros
de la familia.
A partir de 1.992, una nueva técnica mucho más eficaz que la
anterior en la detección de la anomalía es la del uso del método
directo de análisis de ADN, mediante el cual se puede visualizar
la extensión de las repeticiones de las tripletas CGG con
exactitud y así detectar mutaciones y premutaciones. Esta técnica
permite detectar tanto en varones como en mujeres, a individuos
sanos, afectos y portadores, tanto pre como postnatalmente, lo
que permite un asesoramiento genético de gran eficacia.
Las cifras de incidencia en la población
convierten al Síndrome X Frágil en la primera causa hereditaria
de retraso mental y la segunda cromosopatía en frecuencia (después
del Síndrome de Down).
Aunque en nuestro país no existe una estadística
al respecto, se estima que su frecuencia es de 1 varón afectado
por cada 4.000 nacimientos, una hembra afectada por cada 6.000
nacimientos, una portadora por cada 260 y un portador por cada
800. Por ello, globalmente en España puede haber 10.000
afectados por el retraso mental causado por el Síndrome X Frágil
y 100.000 portadores del cromosoma X frágil. Aproximadamente
entre el 80-90% del total falta por diagnosticar.
Hasta ahora se han descrito las causas genéticas
del síndrome X Frágil. Los efectos, que más adelante se
expondrán, son variados siendo el más importante el retraso
mental, que oscila de leve a severo.
Aunque cada persona es diferente, hay una serie
de síntomas comunes que se han observado en personas que padecen
este síndrome.
Los varones afectados por el síndrome X Frágil
tienen como rasgos físicos más característicos macroorquidismo
o testículos grandes (80% de los varones adultos), orejas
grandes y prominentes (80% de los casos), cara larga y estrecha,
mandíbula inferior prominente y problemas de infecciones en el oído
medio.
Aproximadamente un 60% de los varones presentan hipersensibilidad
en las articulaciones. Esto se detecta doblando los dedos hacia
atrás en dirección a los nudillos. También los pies planos
aparecen en un 50% de los niños y adultos.
Otros rasgos son el estrabismo que se detecta en un 25-50% de los
casos y prolapso en la válvula mitral que se presenta en el 80%
de los adultos (consultar ANEXO 1 donde se hace una descripción
más detallada de este problema).
En cuanto a mujeres con síndrome X frágil los síntomas son
semejantes a la de los varones. Las que no se encuentren
afectadas muestran algún rasgo físico propio del síndrome.
La hiperactividad y falta de atención son
problemas de conducta que se presentan tanto en varones como
mujeres afectadas por el síndrome. Lenguaje desordenado y
repetitivo, pobre mantenimiento de los temas, pensamientos
expresados de forma incomprensible son características comunes.
Se han descrito en varones, rasgos que se han calificado como
autistas. Mantenimiento escaso de la mirada, timidez, aleteos con
las manos, repetición de la misma frase constantemente, aversión
a ser tocado o abrazado, rabietas injustificadas, morderse las
manos, se presentan en alrededor del 16% de los varones con X frágil.
Normalmente, los niños X frágil son cariñosos aunque los
rasgos autistas interfieran con la relación normal.
Las mujeres afectadas por el síndrome, suelen presentar leve
retraso mental, problemas de atención pero normalmente sin
hiperactividad. Lo más frecuente es la timidez que puede ser
profunda en la adolescencia, pudiendo llevar a depresión.
En la actualidad, no hay cura para el síndrome
X frágil, aunque se están desarrollando experimentos basados
tanto en terapia genética como en ingeniería genética
consistentes en reproducir la carencia de la proteína causante
de la enfermedad.
No obstante, su tratamiento puede ayudar a los niños a alcanzar
su máximo potencial. Esta ayuda puede ser tanto a nivel médico
para los problemas que anteriormente se han descrito, como
educacionales y de ocupación.
Existen numerosos fármacos que pueden ayudar al control de la
sintomatología, aunque la base de su utilización no es matemática
y hay que "probar" hasta encontrar la que más se
adecue individualmente según la sintomatología que presente, así
como la dosis óptima, e ir revisándola muy de cerca para
adaptarla a los cambios que vayan surgiendo en la patología del
niño y el ambiente en el que se desarrolla.
En ocasiones se utiliza más de una medicación, para tratar una
serie de problemas o por los efectos sinérgicos de las mismas,
los efectos secundarios deben ser monitorizados cuidadosamente
para que no lleguen a superar a los beneficios, también es útil
recordar que la medicación no es el único tratamiento de este síndrome,
como veremos más adelante, pero en la mayoría de los casos,
como en las modificaciones de conducta estas intervenciones
psicológicas son más efectivas con medicación.
Se debe tener en cuenta que como en cualquier síndrome,
en el Frágil X, no todos los rasgos asociados están siempre
presentes en todos los que lo padecen, siendo por tanto necesario
en primer lugar saber las necesidades y habilidades del niño en
concreto. Con frecuencia estos pacientes tienen habilidades para
la imitación, la memoria visual, el humor y son prácticos a la
hora de resolver un problema y aprender. La debilidad más común
es la incapacidad para organizar la información y actuar sobre
la misma de una forma efectiva. Teniendo en cuenta esto, los niños
frágil X, necesitan apoyo en unas áreas determinadas:
- Atención, hiperactividad e impulsividad.
- Aprendizaje.
- Habla y lenguaje.
- Incapacidad para procesar la información sensorial de manera
efectiva y habilidades motoras escasamente desarrolladas.
- Problemas de comportamiento.
Se observan grandes dificultades en el proceso auditivo, procesos
secuenciales, razonamiento abstracto y habilidades aritméticas.
La generalización es difícil y muchas veces una tarea o un
concepto tiene que ser enseñado de varias formas para que el niño
verdaderamente lo aprenda y sea capaz de manejar la información
con sentido.
Las recomendaciones más frecuentes son:
- Control médico para los problemas de
atención y comportamiento.
- Técnicas de autogobierno del
comportamiento que incluyen:
- Fijar la meta, autocontrol, autorreforzamiento y ajuste
de metas.
- Ayuda a los padres a entender los retrasos en el
desarrollo de sus hijos, que en ocasiones es la tarea más
difícil, así como sus comportamientos anormales.
Debemos enseñarlos para que utilicen estrategias para
estructurar el entorno, fomentar y facilitar la producción
de habla y lenguaje, prevenir la sobreestimulación,
utilizar técnicas terapéuticas calmantes y técnicas de
reforzamiento positivo de la conducta.
- Terapia tanto para el habla como para el
lenguaje, así como terapia para desarrollar el
vocabulario y el lenguaje social. Estos niños presentan
lenguaje acelerado, con ritmo desordenado, dispraxia
verbal, articulación pobre, perseverancia, habla
tangencial, falta de sencillez y naturalidad.
- Técnicas de integración sensorial.
Servicios de educación especial, incluyendo un entorno
educativo de apoyo que permita la modificación del
formato instructivo y del plan de estudios.
- Utilizar materiales visuales que el niño
pueda usar para aprender nuevas habilidades y rutinas.
- Utilizar materiales y temas que tengan
gran interés para el niño, y así aprenderá con los
objetos que realmente le gusten, se deben usar además
objetos o fotografías de la vida real y dejar tiempo
para que el niño responda y formule preguntas.
- Hacer que el niño participe en
actividades de pequeños grupos. La imitación es buena
para que adquiera un lenguaje social y un comportamiento
adecuado. Además esta es una cualidad casi constante en
ellos.
- Las dificultades en el proceso auditivo
debe considerarse y la información que se le trasmite al
niño tiene que ser en frases cortas y simples.
- Debemos ir modificando el material pedagógico
para que siempre esté a la altura del desarrollo del niño
y que le dé el apoyo suficiente para que consiga el éxito
por el que está trabajando. La demostración y la
repetición de las áreas son muy útiles para enseñar
nuevas habilidades y rutinas.
Lo más importante de todo es que todas
aquellas personas que estén trabajando con el niño deben
perseguir el mismo propósito, por lo que es fundamental una
coordinación en el trabajo entre los padres, profesores,
psiquiatras y psicólogos, conociendo al niño y aprovechando
todas aquellas cualidades que le pueden ser útiles e intentando
modificar las que le interfieran con un buen funcionamiento psico-social.
Debido a que los síntomas del X Frágil pueden
ser bastante sutiles, especialmente en niños jóvenes y al hecho
que el X Frágil tiene una incidencia notable en la población,
muchos especialistas médicos recomiendan que la prueba sea
tenida en cuenta para cualquier persona con atraso en el
desarrollo o retraso mental de origen desconocido.
El diagnóstico prenatal debe ser realizado a toda aquella
persona en cuya familia se haya detectado algún miembro con
problemas de retraso mental.
El diagnóstico del síndrome de X frágil no se hace por la
presencia de rasgos físicos. Cualquier demostración de retraso
mental, una historia familiar de retraso mental o dificultades de
aprendizaje de etilogía desconocida en combinación con alguna
de las características anteriormente expuestas, pueden hacer
sospechar que nos encontramos con un niño X frágil.
Se quiere resaltar de nuevo el que la única técnica fiable para
detectar este trastorno genético es la que analiza directamente
el ADN. Los estudios citogenéticos ofrecen un porcentaje elevado
de falsos negativos.
Biancalana V.; Taine1 L.; Bouix, J.C. y
col.: Expansion and methylation status at FRAXE can be
detected on EcoRI blots used br FRAXA diagnosis: analysis of four
FRAXE families With mild mental retardation in males. Am J Hum
Genet. Od, 59(4): p847-541 1996.
Carbonell, P.;López, I.; Gabarrón, J y
col.: FRAXE mutation analysis in three Spanish families.
Am. J. Med Genet. 64:434-440,1996.
Castellvi-Bel1 S.; Milá, M.; Soler, A. y
col : Prenatal diagnosis of fragile X syndrome: (CGG)n
expansion and methylation of chorionio vilus samples. Prenat-Diagn.
Sep; 15(9): 801-7,1995.
Hagerman, R J.; Cronister, S: The
fragile X syndrome 2ª edición. University press. 1996.
Hallmayer, J.; Pintado, E.; Lotspeich, L.;
y col.: Molecular analysis and test of linkage between
the FMR-1 gene and infantile autism in multiplex families Am. J.
Hum. Genet. Nov; 55(5): 951-9, 1994.
Hmadcha, A; De Diego, Y.; Pintado, E:
Assesment of Fmr-1 expression by RT-PCR of KH domains. J Lab Clin
Med. 131(2): 170-173,1998.
Mila, M.; Castellvi-Bel, S; Sánchez, A;
Lazaro. C; Villa M.; Estivill, X.: Mosaicism for the
fragile X syndrome full mutation and deletions within the CGG
repeat of the FMRI gene. J Med Genet Apr. 33(4): p338-40, 1996.
Pascual Pascual, S.I.; Garcia Marcos, J.A.;
Martin Lucas, M.A.: Estudio del síndrome X-frágil en la
población de la compañía telefónica de España. Rev. Neurol.
May-Jun; 23(121): 644-7, 1995.
Pintado, E; de Diego, Y.; Hmadcha, A.;
Carrasco1 M.; Sierra, J.; Lucas L.: Instability of the
CGG repeat at the FRAXA locus and variable phenotypic expression
in a large fragile x pedigree. J. Med. Genet 32: 907-908, 1995.
Turner, G.; Webb, T.; Wake, S.; Robinson,
H.: Prevalence of fragile X syndrome. Am J Med Genet. Jul
12,64(1): p 196-7, 1996.
Willemsen, R.; Mohkamsing, S.; De Vries,
B.; y col.: Rapid antibody test for fragile X syndrome
Lancet 345: 1147-1148,1995.
Aguirre, A. y Femández-Rua, J. M.:
"La neurociencia proyecta rayos de luz sobre numerosos desórdenes
del cerebro". ABC de la Ciencia 27 de marzo de 1998 -
Borrero, J: ABC de la Sanidad. ABC de Sevilla. 10 de Abril de
1997.
Carrasco, M., Pintado, E. Lucas, L., M De
Diego, Y. Hmachda, A.: Intentemos prevenir lo que hoy no
podemos curar: La deficiencia mental ligada a la fragilidad del
cromosoma X. Revista Clave. Real patronato de prevención y
atención a personas con minusvalías. Madrid 1996
De Diego Y.: Síndrome X Frágil
Edición de bolsillo. Libro publicado por I Jornada Andaluza
sobre el Síndrome X Frágil. Sevilla, 13 Julio 1998.
López. M.: El Síndrome del X Frágil:
La causa más común de subnormalidad en el Hombre. Mundo Científico,
Nº. 141, vol 13: 1066-1068.
Pintado E., de Diego Y., Hmadcha A., y
Lucas M.: Diagnóstico Molecular del Síndrome X Frágil.
Capítulo 10 del libro Diagnóstico Molecular en Genética Médica.
Sociedad Española do Bioquímica Clínica y Patología Molecular.
Dirigido por Lucas. M (1998).
Ramos, F. J: EI Síndrome X Frágil
Material educativo de la Fundación Nacional del X Frágil de
Estados Unidos". Editado por IMSERSO 1998 Número 53.
Colección Rehabilitación.
Ruiz. M J. y Gómez-Ferrer, C.:
Revisión de los tratamientos en el Síndrome Frágil X. Revista
Electrónica de Psiquiatría Vol 1, Nº. 4. Diciembre 1997.
(Fuente: Libro El Síndrome X Frágil. Edición
de bolsillo. Publicado para la I Jornada Andaluza del Síndrome X
Frágil. Editado por la doctora Yolanda de Diego Otero
A: abreviatura de Adenina
Adenina: Base nitrogenada que forma parte del ADN y
del ARN.
ADN: Ácido desoxirribonucleico, se encuentra
principalmente formando parte de los cromosomas Es la molécula
en la que está codificada la información genética.
ADNc: Ácido desoxirribonucleico complementario de
un ARNm
ADN genómico: ADN que forma parte de los
cromosomas.
ADN repetitivo: Secuencia de longitud variable que
se repite un número de veces alto en el genoma.
Alelo: Nombre que se le da a dos formas distintas
de un mismo gen en los cromosomas homólogos.
Alelos múltiples: Cuando aparecen mas de dos
formas de un gen concreto en la población.
Aminoácido: Molécula que posee un grupo carboxilo
y uno amino, forma parte de todas las proteínas
Amniocentesis: Procedimiento usado en el diagnóstico
prenatal para obtener amniocitos.
Amniocitos: Células de origen fetal que se
encuentran en el liquido amniótico.
Anticipación: Tendencia observable, en algunos
tipios de enfermedades en las que los síntomas aparecen mas
graves y precoces en generaciones sucesivas.
Autosomas: Cromosomas que aparecen en las células
del organismo y son distintos a los sexuales, en los humanos
aparecen 22 pares en cada célula somática.
Base nitrogenada: componente de los ácidos
nucleicos, en los que aparecen cinco tipos distintos de bases:
Adenina, Citosina, Guanina, Timina y Uracilo
Biopsia de vellosidades coriónicas: Procedimiento
utilizado en el diagnostico prenatal para obtener una muestra de
tejido fetal.
C: Abreviatura de Citosina
Carácter: Característica o rasgo que se observa
en un individuo y forma parte de su fenotipo
Célula: Unidades básicas de las que se componen
los organismos pluricelulares.
Célula germinal: Aquellas células del organismo
que transmiten la información genética a la siguiente generación
de individuos.
Cigoto: Óvulo que ha sido fecundado por un
espermatozoide.
Citoqenética: Rama de la genética que se ocupa
del estudio de los cromosomas.
Citoplasma: Parte de la célula limitada por la
membrana externa y la nuclear.
Congénita: Anomalía presente en el individuo a
nacer.
Cromosomas: Estructuras filiformes que se observan
en los núcleos de las células en el momento de la división
celular están formados por ADN y proteínas, en ellos está
codificada la información genética que sirve para determinar
todas las funciones y formas de las células de un organismo vivo.
Cromosomas sexuales: Par de cromosomas distinto a
los pares autosómicos, que están implicados, entre otras
funciones, en la determinación del sexo del organismo, en la
mujer son dos cromosomas X, y en el varón un X y un Y.
CpG: Región o isla de ADN rica en citosinas y
guaninas, regula la transcripción de los genes eucariotas, entre
ellos el gen Fmr-1.
Delección: Pérdida de un fragmento cromosómico.
Diagnóstico en la preimplantación: Detección de
un trastorno hereditario antes de la implantación, realizada en
un cigoto tras una fecundación "in vitro".
Diagnóstico prenatal: Pruebas empleadas para
determinar si el feto presenta alguna alteración que e provoque
alguna enfermedad.
Dominante: Carácter que se manifiesta siempre en
los individuos aunque los dos alelos el gen o genes que lo
determinan sean diferentes (heterocigotos).
Dosis génica: Cantidad total de genes de un
individuo.
Efecto fundador: Efecto observado en algunas
enfermedades genéticas, en las que los individuos afectados en
la actualidad, descienden de un antepasado común.
Fenotipo: Características físicas, bioquímicas y
fisiológicas de un individuo que resultan de la interacción del
genotipo y el entorno.
Fmr-1: Gen del retraso mental ligado al sitio frágil
1, situado en el locus FRAXA.
Fmr-2: Gen del retraso mental ligado a sitio frágil
2, situado en el locus FRAXE.
G: Abreviatura de Guanina, base nitrogenada
componente de ADN.
Gameto: Célula germinal del organismo puede ser óvulo
o espermatozoide y contiene una sola copia de cada cromosoma (dotación
genética haploide).
Gen: Parte de la secuencia del ADN que codifica
para una secuencia de aminoácidos en la proteína
Genoma: Conjunto de todos los genes de una célula.
Genotipo: Constitución genética de un individuo.
Haplotipo: Variantes que se observan en una región
cercana, ligada a un gen o alelo concreto, del mismo cromosoma.
Heterocigoto: Individuo que muestra dos alelos
distintos en un gen concreto en el par de cromosomas homólogos.
Homocigoto: Individuo que muestra dos alelos idénticos
en un gen concreto en el par de cromosomas homólogos.
Inactívación del cromosoma X: Proceso que ocurre
en las células femeninas debido a que llevan dos cromosomas X,
para compensar la dosis génica respecto a las células
masculinas que sólo llevan un cromosoma X.
Inestabilidad cromosómica: Punto de rotura del
cromosoma que se asocia a ciertas patologías o a alteraciones en
la secuencia de nucleótidos.
"In vítro»: En el laboratorio (literalmente
en cristal).
IQ: coeficiente de inteligencia (valor numérico).
Ligamiento: Se dice que dos genes están ligados
cuando están tan cerca en el cromosoma que se observa que se
heredan juntos.
Ligamiento al cromosoma X: Genes situados en el
cromosoma X
Ligamiento al sexo: Caracteres que se encuentran
determinados por genes situados en los cromosomas sexuales X o Y.Locus
(Plural Loci): Lugar en el que se sitúa un gen en un
cromosoma.
Macroorquidismo: Tamaño de los testículos por
encima de la media de la población.
Mosaicísmo: Presencia de distintos caracteres genéticos
en los tejidos del organismo.
Mutación: Cambio ocurrido en el material genético
que se hereda.
Núcleo: Estructura de las células eucariotas
donde se sitúan los cromosomas.
Nucleótido: Unidades básicas del ADN formada por
una base nitrogenada, una pentosa y un grupo fosfato, sirven de códigos
para los caracteres heredables.
POR: Reacción en cadena de la polimerasa, se
consigue amplificar parte del ADN.
Penetración: Proporción de individuos
heterocigotos para un carácter en los que se manifiesta aunque
sea levemente.
Portador obligado: Individuo que por el análisis
de su familia tiene que tener el carácter que se analiza.
Prevalencia: Cálculo de la proporción de
individuos con un carácter en una población.
Proteína: Molécula biológica que esta formada
por cadenas de aminoácidos, y que determina una función celular.
Síndrome: Conjunto de características o síntomas
que aparecen juntos en una patología o enfermedad
"Southern-blot": Detección en los
polimorfismos de restricción del ADN por hibridación con sonda
marcada.
T: Abreviatura de timina.
Telméro: Parte final de un cromosoma.
Terapia genética: Tratamiento de una enfermedad
introduciendo el gen alterado en las células del organismo vivo.
Traducción: Proceso por el que un ARNm es
interpretado como una secuencia de aminoácidos de una proteína.
Triplete: Secuencia de tres nucleótidos en la molécula
de ADN o en el ARNm y que codifica para un aminoácido concreto
en la proteína.
Xq 27.3-28: Bandas finales del brazo largo del
cromosoma X.
(Fuente: Libro El Síndrome X Frágil. Edición
de bolsillo. Publicado para la I Jornada Andaluza del Síndrome X
Frágil. Editado por la doctora Yolanda de Diego Otero
Este trabajo ha sido realizado por
la doctora Esther
Rodríguez Adradal.
Según datos de estudios realizados, un porcentaje elevado de
adultos que padecen el Síndrome X Frágil pueden tener
complicaciones cardiológicas en la válvula mitral.
1 - Configuración y
funcionamiento cardiaco.
EI corazón es el órgano
central del aparato circulatorio, formado por músculo y
semejante a una bomba impulsora. En realidad está constituido
por dos bombas separadas por un tabique, dividiendo así al corazón
en dos partes laterales.
- Corazón derecho, por el que circula la sangre sin oxígeno (venosa)
- Corazón izquierdo, en relación con sangre oxigenada (arterial)
A su vez, cada una de estas des mitades laterales se subdividen
en otras dos, una superior llamada aurícula y otra inferior o
ventrículo.
Ahora bien, así como los dos corazones están enteramente
separados uno del otro, cada una de las aurículas comunica
ampliamente con el ventrículo correspondiente al mismo lado por
medio de un ancho orificio denominado auriculo-ventricular
provisto de una aparato mecánico llamado válvulas, que en el
corazón izquierdo se denomina mitral y en el derecho tricúspide,
regulando el curso de la sangre. Estas descienden en el momento
de la relajación cardiaca, dejando, que la sangre pase la aurícula
al ventrículo, y se elevan durante la contracción cardiaca para
evitar que esta misma sangre vuelva a ascender hasta la aurícula.
(T1)
2. Circulación.
El ventrículo izquierdo bombea
sangre arterial penetrando en la arteria Aorta, la cual es la que
se encarga de distribuirla a todos los órganos del cuerpo, cediéndoles
oxigeno y tomando de ellos sustancias de desecho transformándose
así en sangre venosa. Ésta a través de las venas llega a la
aurícula derecha y pasa al ventrículo derecho por la válvula;
el ventrículo derecho lo impulsa a la arteria pulmonar, llegando
la sangre hasta los pulmones donde se despoja de su ácido carbónico
y se carga con oxigeno, volviendo a tomar camino hacia el corazón,
introduciéndose en la aurícula izquierda y por medio de su válvula
al ventrículo izquierdo comenzando de nuevo el ciclo. (T2).
3. Prolapso de la válvula mitral.
Síndrome clínico que afecta
frecuentemente a personas entre los 15 y 30 años. En la mayoría
de los pacientes se desconoce la causa, asociado también a otras
enfermedades en las que existe alteraciones del tejido conectivo.
Los pacientes portadores de prolapso de válvula mitral,
presentan un desplazamiento anormal de una o ambas valvas de la válvula
mitral (normalmente la valva posterior) hacia la aurícula
izquierda durante la contracción del ventrículo, permitiendo
que pequeñas cantidades de sangre fluyan hacia la aurícula,
produciendo durante la exploración física que el médico
realiza al enfermo lo que se denomina soplo. Esto se debe a que
los músculos papilares que sustenta las valvas son demasiados
largos e hipertrofiados.
La mayoría de estos pacientes no pero cuando se produce alguna
manifestación los clasificamos en:
- Cardiacas: Dolor en el tórax que se localiza
retroesternalmente sobre todo en reposo. Palpitaciones o sensación
de que el corazón va a salirse de la caja torácica. Mareo. Síncope.
Disnea o falta de aire.
- Extracardiacas: Convulsiones. Hemiparesias. Ambas consecuencia
de falta de riego sanguíneo transitorio. Cefaleas. Crisis de
ansiedad. Trastorno de pánico. Depresión. Hiperactividad.
4 - Complicaciones del prolapso de
la válvula mitral.
- Insuficiencia mitral, siendo
el prolapo de la válvula mitral la principal causa de esta
insuficiencia.
- Infección del endocardio.
- Muerte súbita, complicación grave pero muy rara.
5 - Exploración física.
Como comentamos anteriormente el
soplo sistólico constituye el dato más característico a la
auscultación. La tensión arterial habitualmente no suele estar
alterada. Si bien, es posible observar alteraciones musculoesqueléticas
(rectificación de curvas fisiológicas de la columna, pecho en
paloma). Como métodos complementarios al diagnóstico se usa el
electrocardiograma, ecocardiograma y angiografía.
6 - Tratamiento
- Si no presenta síntomas, no
es necesario realizar nada.
- Tratamiento sintomático del dolor torácico.
- Si existe insuficiencia mitral se debe realizar una revisión
cada 6 o 12 meses, dependiendo del grado de alteración
cardiaca que presente el caso. Si la insuficiencia mitral
fuese grave y se asociase a alteraciones hemodinámicas estaría
indicada la cirugía (sobre todo sustitución valvular).
- Profilaxis frente a la infección del encocardio, solo si
se evidencia alteración de las valvas (engrosamiento). Y
cuando se proceda a:
- Manipulación buco-dental.
- Técnicas sobre el tubo digestivo y vías genitourinarias.
- Cirugía cardiaca.
En el momento actual y promovido
por la necesidad de encontrar algún tipo de terapia que permita
un tratamiento efectivo del síndrome, se están realizando en
casi todos los países desarrollados, programas de investigación
básica y aplicada sobre el síndrome. Uno de los objetivos
principales de la investigación es conseguir una terapia de
sustitución proteíca o genética.
Con fondos gubernamentales en los
países desarrollados y con ayudas de las Fundaciones
Norteamericanas para la investigación del Síndrome X Frágil,
se mantienen programas científicos para desarrollar posibles
terapias, que palíen los efectos dramáticos del síndrome en
las personas que lo padecen. Tratan sobre modelos no humanos del
síndrome en los que la investigación básica pueda realizarse
sin dificultad, como son el modelo de levadura o el ratón nulo
del gen Fmr-1. En este último se realizan importantes esfuerzos
para identificar las posibles alteraciones del cerebro cuando la
proteína FMRP está ausente en sus células.
Otra línea abierta es la
investigación de la función de la proteína en la sinapsis
neuronal y en la regulación celular, y el estado de metilación
del promotor del gen Fmr-1. También se estudia el uso de
AMPAkinas en el tratamiento de la memoria en estos pcientes, así
como el uso de nuevos fármacos que puedan tratar eficazmente los
síntomas en los pacientes afectados por el síndrome.
La frecuencia de un defecto genético
grave en los recién nacidos es 1 de cada 100. De los 4000
trastornos hereditarios que se conocen, hay muchos en los que la
terapia farmacológica es ineficaz. La tendencia actual es
investigar la manera de introducir el gen o una proteína
funcional en las células somáticas del individuo afectado por
la enfermedad, sin afectar las células germinales, por lo que la
posibilidad de que el gen manipulado a las siguientes
generaciones es nula.
En las enfermedades en las que el
tejido afectado es el neuronal, como es el caso del Síndrome X
Frágil, la actuación se complica, debido a que dirigir la
terapia al sistema nervioso central es difícil por la
complejidad estructural y funcional del tejido, por las barreras
naturales que muestra y por el insuficiente conocimiento que se
tiene aún sobre el sistema nervioso. En principio, la terapia génica
puede ser no solo un tratamiento sino una cura radical de la
enfermedad. Sin embargo será necesaria mayor investigación para
conseguir superar los problemas que puede presentar el uso de la
terapia génica en el Síndrome X Frágil y queda abierta la
posibilidad de curación de los pacientes actuales o de aquellos
que nazcan en el futuro.
(Fuente: Libro El Síndrome X Frágil. Edición
de bolsillo. Publicado para la I Jornada Andaluza del Síndrome X
Frágil. Editado por la doctora Yolanda de Diego Otero
El éxito de las distintas
estrategias que pueden utilizarse para el tratamiento de los síntomas
observados en el síndrome, depende de la dedicación,
creatividad y flexibilidad de un equipo multidisciplinar que
trabaje coordinadamente para tratar a estos pacientes. Los
padres, educadores, terapeutas, psicólogos y médicos juegan un
papel muy importante a la hora de determinar los objetivos académicos
y terapéuticos para cada niño. Los individuos con el Síndrome
X Frágil necesitan una intervención individualizada que tenga
en cuenta las necesidades de cada paciente particular. Por lo
tanto será necesario un abordaje integrado en la intervención y
educación de cada paciente, que combine la experiencia de
padres, médicos, doctores, genetistas, psicólogos, pedagogos,
logopedas, terapeutas ocupacionales, profesores de educación
especial y otros profesionales.
Por las características de los
pacientes con este síndrome, esta patología requiere la
intervención médica y terapéutica como componentes de un
servicio integral. Se necesitan estrategias específicas para las
distintas edades de los individuos con el síndrome, que
comprendan la infancia, edad escolar, adolescencia y edad adulta.
En los niños con el síndrome se
observan problemas de atención, dificultades para resolver algún
problema concreto, así como para entender conceptos abstractos y
memorizar o aplicar información conocida en nuevas situaciones.
En los programas más extendidos en países como Estados Unidos,
Australia o algunos países europeos, se realiza, desde finales
de los años 80 una intervención integrada e individualizada
para cada familia afectada por el síndrome, haciendo cambios en
los tratamientos educacionales, servicios relacionados e
intervención terapéutica y médica, intentando tratar a estos
pacientes en el ambiente menos restrictivo posible. Estudios
sobre la efectividad de los tratamientos en estos pacientes
indican que aumenta cuando se realiza en medios y contextos
funcionales comparándolos a los realizados en medios aislados.
A principios de los años 90, en
Estados Unidos, se amplían los objetivos para personas con
minusvalías, integrándolas todo lo posible en los aspectos
normales de la sociedad, incluyendo programas educativos y
espacios comunitarios. Se hace énfasis en los servicios de
asistencia técnica y en la adaptación y transición de la fase
escolar a la integración laboral y social completa. Se prohibe
la discriminación de niños y adultos discapacitados en los
empleos públicos y privados, así como otros servicios y
acomodaciones, integración que esta incluida en los planes y
acciones sobre minusvalías en España. Los servicios integrados
adaptados al Síndrome X Frágil, deben tener en cuenta las
complejas necesidades que se observan en los pacientes, entre las
que se encuentran: la hipersensibilidad a los estímulos
sensoriales, visuales, sonoros, olfativos y de movimientos, así
como una reacción desmesurada ante novedades.
Presentan dificultades en la
transición y los cambios, dificultades en las funciones
reguladoras entre las que están los problemas para mantener la
atención, la modulación de los niveles de actividad y la
impulsividad. También muestran dificultades en las capacidades
oralmotoras, motora gruesa y fina y visuomotora; problemas en las
habilidades sociales y del lenguaje; dificultades en el habla,
como la rapidez, los desórdenes en el ritmo, dispraxias
verbales, haciendo en muchos casos que el habla sea
incomprensible. Se pueden citar otras características como déficit
cognitivo que va desde problemas de aprendizaje a retraso mental
severo, lo que conduce a limitaciones importantes en el
desarrollo de la vida diaria.
En cada caso concreto es
recomendable hacer un programa individual de actividades diarias,
que tenga en cuenta la estimulación precoz en los casos de niños
menores de 3-4 años, y en los mayores una terapia ocupacional
con un enfoque sensorial e integrativo en una terapia logopédica,
o también una estrategia que combine las dos terapias
anteriores, así como una intervención educativa y de
comportamiento adaptativo en los niños en edad escolar.
También tendrá que tenerse en
cuenta la posibilidad de modificaciones en el ambiente o entorno
habitual del niño con el síndrome, siendo éste lo más
flexible y cómodo para conseguir un desarrollo integral, se
realizará un control de la medicación y un descanso adecuado.
Se recomienda usar técnicas de relajación; se eliminarán
posibles factores de distracción, teniendo en cuenta la
luminosidad ambiental, el ruido, los olores, el uso de juguetes
adecuados, etc. Se realizarán los exámenes y evaluaciones en el
momento más adecuado para el niño y se mantendrá un contacto
estrecho con la familia, siendo el programa flexible, abierto y
creativo.
Los pacientes con el síndrome
pueden mostrarse extremadamente agitados en los momentos de
transición entre actividades debido a la dificultad de modulación
sensorial, por lo que debe explicarse al niño claramente que va
a ocurrir en cada momento, ya sea verbalmente, con esquemas,
fotografías o dibujos. Es útil revisar el esquema diario a través
de fotografías, usar canciones o músicas para anunciar los
cambios, así como cambios de luces o sonidos armónicos, para
mejorar el proceso de adaptación; también pueden usarse
juguetes concretos con los que el niño esté habituado, para
iniciar las distintas actividades.
Debe tenerse en cuenta en estos
pacientes la posibilidad de realizar actividades que calmen su
agitación y ansiedad, como pueden ser: colocar al paciente entre
mantas, almohadas o cojines suaves y cómodos, entre capas de
neopreno o llevar guantes de este material. También pueden
realizar trabajos simples o repetitivos como llevar libros,
alguna caja, subir escalones, montar en bicicleta o colocarse
contra la pared. Se pueden realizar movimientos rítmicos,
escuchar sonidos vibrantes o música y ritmos relajantes, como
una rutina diaria para reforzar el aprendizaje. Se pueden usar técnicas
de yoga y respiración, así como el tiempo de silencios como
parte del proceso de aprendizaje.
Realizar actividades en comunidad
permite a estos individuos llevar un estilo de vida mas calmado y
equilibrado. Pueden cuidad animales domésticos, montar a
caballo, realizar artes marciales, ayudar en tareas domésticas o
en granjas. Es muy beneficioso relacionarse con otros niños en
zonas de juegos como parques o visitar museos. Por todo lo
expuesto, en estos pacientes se determinarán las actividades de
manera individual siguiendo un programa de adaptación y
aprendizaje concreto que permita la adquisición de nuevas
capacidades o la ampliación de las ya adquiridas por el
individuo afectado.
Propuesta de intervención en
el Síndrome X Frágil
En la actualidad se están
llevando a cabo en muchos grupos de tratamiento del síndrome un
tipo de enfoque multidisciplinar coordinado. Con este abordaje se
pretende conseguir un tratamiento efectivo de las personas
afectadas por esta patología, consiguiéndose mejorías
apreciables en la adaptación social y laboral de estos pacientes.
Este enfoque involucra una serie
de actuaciones que se van a exponer a continuación.
1 - Valoración clínica
Una valoración clínica adecuada
es fundamental para detectar a los posibles individuos afectados
con el Síndrome X Frágil. En la actualidad es el primer paso de
una intervención médica efectiva. Por ello será necesario un
esfuerzo inicial para informar a los facultativos que serán los
primeros que entren en contacto con los individuos afectados. Es
necesario que los médicos de familia, pediatras y neuropeditras
conozcan muy bien las características que muestran estos
pacientes y la variabilidad de síntomas que pueden ser debidos
al síndrome. Una valoración clínica precoz de estos individuos
permitiría una intervención adecuada en el desarrollo
psicomotor del paciente, que le permita alcanzar el máximo nivel
posible.
2 - Diagnóstico genético
molecular
Una vez se ha realizado la
valoración clínica del paciente y si se sospecha que puede
padecer el Síndrome X Frágil, se deberá descartar por pruebas
genéticas moleculares. Debido a la alta prevalencia de este síndrome
(1 individuo afectado por cada 2500-4000) y a las múltiples
consecuencias, será necesario poner a punto las técnicas de
diagnóstico en los laboratorios de genética y biología
molecular de los servicios sanitarios, lo que permitiría un
diagnóstico con certeza el Síndrome. Es también necesario que
existan unidades de diagnóstico prenatal efectivas que permitan,
de forma rápida y fiable, descartar el síndrome en los
embarazos de riesgo.
Es necesario realizar un diagnóstico
preciso y rápido del síndrome que permita detectar a las
familias con el síndrome, lo que permitirá llevar a cabo el
siguiente paso de este modelo de intervención y evitar a su vez
situaciones de estrés en las familias afectadas.
3 - Consejo genético y
asesoramiento familiar
Detectada ya la familia afectada,
por el estudio de un individuo con el síndrome, será
conveniente analizar el resto de individuos que tengan riesgo de
haber heredado el cromosoma X alterado (ya sea con la premutación
o la mutación completa). En algunos casos podrán detectarse
portadores sanos (sin rasgos clínicos de la patología) pero que
tienen un probabilidad alta de tener hijos o nietos afectados.
Es necesario explicar el patrón
hereditario y el tipo de análisis o pruebas que tendrán que
realizarse a los miembros de la familia. El consejo genético
permite conocer el riesgo empírico que existe de heredar el
cromosoma X alterado y aconsejar a la familia sobre las
posibilidades que existen en la actualidad para tener
descendencia sana, realizando un enfoque preventivo de la patología.
Entre las posibilidades actuales se encuentran el diagnóstico
prenatal (amniocientesis, vellosidades conrionicas o sangre fetal),
diagnóstico preimplantacional se realizan las pruebas
moleculares en los embriones obtenidos por fertilización "in
vitro" y solo se implantan aquellos que no hayan heredado el
cromosoma mutado. También se puede recurrir a un donante de
gametos o la adopción de niños ya nacidos. Todas estas
posibilidades permiten a las familias decidir entre ellas a la
hora de tener otros hijos.
4 - Seguimiento médico
especializado
Una vez que se ha detectado el síndrome
en una familia, será conveniente realizar un seguimiento por
parte del médico de familia o especialista correspondiente, que
permita abordar el tratamiento de los múltiples síntomas que
suelen aparecer en este síndrome, entre ellos: otitis, paladar
hendido, hernias, anomalías cardiacas, dificultad respiratoria,
hiperactividad, hipotonía y retraso motor. Aparición de timidez
o ansiedad excesivas así como otro tipo de alteraciones
cognitivas. Muchos de estos síntomas se tratan de manera
efectiva con fármacos o terapias, ello evita mayores secuelas en
los pacientes, como pueden ser la falta de audición, problemas
de comportamiento y aislamiento, así como problemas de lenguaje
y aprendizaje.
Será necesario a su vez valorar
el desarrollo psicomotor del paciente a través de un examen
exhaustivo tanto físico como nerurológico, realizándose un
tratamiento adecuado de aquellos síntomas que pueden favorecerse
con terapias disponibles en la actualidad.
5 - Estimulación precoz
Para intentar minimizar las
alteraciones cognitivas provocadas por el síndrome en los
individuos afectados, se puede seguir un programa de estimulación
precoz que permitirá desarrollar y ampliar sus capacidades y
habilidades al máximo. Para que sean eficaces deben iniciarse en
los primeros años de vida. Este tipo de enfoque terapéutico
puede llevarse a cabo de manera coordinada con la familia, que
puede realizar una labor importante en la estimulación precoz
del individuo afectado.
6 - Tratamiento farmacológico
En la actualidad no se ha
desarrollado un tratamiento curativo para el síndrome que
permita recobrar el estado normal en los pacientes nacidos con
esta enfermedad, pero si hay fármacos que tratan de forma
efectiva muchos de los síntomas descritos, aliviando y
permitiendo en muchos casos llevar una vida familiar y social más
normalizada.
7 - Logopedia, terapia del
habla y del lenguaje
Las alteraciones en el lenguaje y
habla de los pacientes con el síndrome son rasgos muy frecuentes.
En los varones se suele observar mayor velocidad, ritmo irregular
y dispraxia verbal (falta de planificación motora del habla).
Para conseguir una pronunciación adecuada es necesario realizar
movimientos finos suaves y rápidos con la lengua y los labios.
Esta terapia intentará conseguir que el paciente desarrolle la
capacidad de hablar a través de estímulos visuales, auditivos y
motores, presentando la información como una imagen única que
será más fácil de aprender para este tipo de pacientes.
El rendimiento suele disminuir a
medida que el lenguaje adquiere estructuras y conceptos
abstractos, pero estos pacientes tienen muy desarrollada una
capacidad alta de imitación verbal. La verborrea, los
comentarios evasivos y el contacto visual escaso, serán otros
problemas que deban tratar el logopeda, teniendo siempre en
cuenta la dificultad de adaptación a nuevos ambientes en estos
pacientes, por lo que el tratamiento será difícil hasta que el
paciente se adapte a los nuevos cambios y a la terapia.
8 - Integración sensorial,
tratamiento psicopedagógico y conductual
Debido a la variabilidad en los síntomas
de estos pacientes, es necesario conocer las necesidades y
habilidades de cada niño concreto. Normalmente es necesario un
apoyo en áreas como la atención, la hiperactividad, la
impulsividad, la concentración, la relajación o las habilidades
sociales. Será necesario conseguir un procesamiento de la
información sensorial de manera efectiva, así como un
desarrollo de las capacidades motoras finas llevado a cabo por un
fisioterapeuta.
Es recomendable un tratamiento de
los problemas de comportamiento por el especialista, así como
una colaboración estrecha con la familia para conseguir
estructurar el entorno, prevenir la sobreestimulación, fomentar
el lenguaje, conseguir el uso de técnicas calmantes y de
reforzamiento positivo de la conducta.
9 - Plan educativo
individualizado
Todas las personas que están
trabajando en la terapia y aprendizaje del niño afectado por el
síndrome, deben perseguir el mismo propósito. Para ello sería
fundamental una coordinación terapéutica, educativa y familiar.
En la etapa escolar es beneficioso estar en un colegio de
integración por la capacidad de aprendizaje por imitación. Es
bueno no sobreestimular su deficiente desarrollo cognitivo con
ambientes excesivamente exigentes, pues su velocidad de
aprendizaje es menor que en el resto del grupo y suelen llegar a
un limite de desarrollo.
El apoyo psicopedagógico es
fundamental en estos niños para conseguir desarrollar al máximo
las potenciales individuales y corregir aquellas conductas que
interfieren en el aprendizaje. Por ello sería útil que
asistiesen a clases de educación especial dentro de su programa
educativo integrado. También puede ser muy útil el uso de las
nuevas tecnologías, como la informática, para mejorar el
desarrollo cognitivo con programas informáticos específicos.
10 - Terapia ocupacional y
asistencia sociolaboral de adultos
Conseguidas unas metas de
aprendizaje de concepto en estos pacientes y debido a sus
limitaciones intelectuales, es posible continuar el aprendizaje
en un taller de terapia ocupacional, en los que podrán
desarrollar una actividad productiva satisfactoria debido a sus
buenas habilidades motoras. En muchos de estos pacientes es muy
útil realizar una integración sensorial junto con la terapia
ocupacional, lo que favorece su relajación y concentración
Ayudar en tareas de transporte de
material pesado, realizar tareas mecánicas y repetitivas
rutinarias es muy útil para reducir las tensiones que acumulan.
Es necesario un apoyo familiar y asistencial para conseguir una
integración social y laboral en estos pacientes. Estos esfuerzos
permiten a las personas sentirse útiles y desarrollar todas sus
capacidades al máximo.
CONCLUSIONES
Este enfoque propuesto a lo largo
de los diez puntos expuestos, definido como tratamiento
multidisciplinar integrado, requiere a su vez la existencia de
especialistas, ya sean en centros públicos o privados, que estén
directamente involucrados en conseguir normalizar en lo posible
las vidas de estos pacientes, que ya al nacer presentan una serie
de necesidades muy amplias, para las que en muchos casos se
dispone de tratamientos específicos en la actualidad.
A su vez, se necesita una
colaboración esencial de las familias afectadas para que el
tratamiento sea efectivo. Su esfuerzo junto con el de los muchos
especialistas involucrados en el tratamiento multidisciplinar,
debe perseguir un tratamiento eficaz y duradero, que permita una
integración social y laboral de cualquier persona que nazca con
este tipo de discapacidad psíquica conocida con el nombre de Síndrome
X Frágil.
La situación del Síndrome X Frágil
en España puede resumirse en los siguientes puntos:
- Esta enfermedad es muy
frecuente y afecta a grandes familias, en las que pueden
aparecer individuos afectados a lo largo de distintas
generaciones, tanto varones como hembras. Por los datos
de prevalencia calculados faltan aún muchas familias con
el síndrome por diagnosticar, aproximadamente el 80-90%
del total. Usando los datos de prevalencia de 1 individuo
afectado por cada 4000 y una portadora por cada 800 y un
portador por cada 5000, se estima que existen unos 8000
varones portadores normales, 10000 individuos afectados
por el retraso mental causado por el Síndrome X Frágil
y 50000 mujeres portadoras del cromosoma X alterado. En
muchos de los portadores sanos tienen una posibilidad altísima
de tener hijos o nietos afectados por el Síndrome X Frágil.
- La falta de conocimiento de
este síndrome plantea la necesidad de una divulgación
efectiva de este tipo de retraso mental hereditario a
todos los niveles:
- Profesionales: médicos
de familia, especialistas, profesores, psicólogos.
- Instituciones y
estamentos públicos.
- Población general.
- Las familias afectadas por el
síndrome necesitan una atención médico-profesional
efectiva por lo que será necesario:
- Equipos de diagnóstico
eficaces.
- Fiabilidad en la
detección prenatal y postnatal.
- Información y
consejo genético adecuado.
- Asistencia psicológica.
- Atención
especializada integrada a distintos niveles.
- Estimulación precoz.
- Terapia ocupacional.
- Logopedia, pedagógica
y psicoterapia.
- Existe una necesidad clara de
diagnóstico precoz en los individuos nacidos son el síndrome
y de consejo genético a las familias con individuos
afectados, todo ello dirigido a detectar los portadores
normales de la población, que tienen riesgo de
transmitir el síndrome a sus descendientes. Esta
necesidad se justifica por el importante coste personal,
familiar y social del Síndrome X Frágil.
(Fuente: Libro El Síndrome X Frágil. Edición
de bolsillo. Publicado para la I Jornada Andaluza del Síndrome X
Frágil. Editado por la doctora Yolanda de Diego Otero